Features
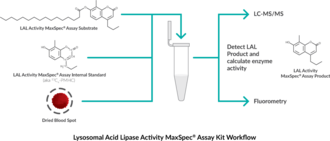
- Easy-to-use reagent kit for the quantification of lysosomal acid lipase activity in dried blood spots
- Designed for use in LC-MS-based or fluorometry applications
- Includes necessary substrate, product, and internal standard, all provided at known concentrations
Technical Support & Resources
Visit our FAQ
Contact Us
Toll Free Phone (USA and Canada Only): (888) 526-5351
Direct Phone: (734) 975-3888
Product Categories
Product Type
Lysosomal Acid Lipase Activity MaxSpec® Assay Kit
Item No. 24854
Technical Information
- LAL
Shipping & Storage Information
Recommended Products
Certificates of Analysis & Batch Specific Data
Provide batch numbers separated by commas to download or request available product inserts, QC sheets, certificates of analysis, data packs, and GC-MS data.
Lipid Resource Center
Discover Products & Resources for Lipid Research
- High-purity lipid standards
- Lipid roles in biology
- Lipids in health & disease
- Lipids for pharmaceutical development
- Protocols, advice, & resources
Product Description
Lysosomal acid lipase (LAL) is a lysosomal enzyme that hydrolyzes cholesteryl esters and triglycerides to produce cholesterol, glycerol, and free fatty acids.1 LAL deficiency is due to mutations in the LAL gene, LIPA, that lead to decreases in LAL activity. Wolman's disease is a severe form of LAL deficiency that begins in infancy and is characterized by a nearly complete or complete lack of LAL activity resulting in gastrointestinal disorders, hepatomegaly, and failure to thrive, leading to hypercholesterolemia and fatality within months without treatment. Cholesterol ester storage disorder is a less severe form of LAL deficiency in which LAL activity is reduced but not abolished. It presents later in life and is characterized by gastrointestinal disturbances, dyslipidemia, hepatomegaly, and impaired liver function. Research towards development of methods to detect deficiency in this enzyme has become an important goal in diagnosing and treating individuals with this disorder.
WARNING This product is not for human or veterinary use.
References & Product Citations
1. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2(9), 670-679 (2017).
LC-
Contact Info
Cayman Chemical1180 East Ellsworth RoadAnn Arbor, Michigan 48108 USAToll Free: (800) 364-9897(USA and Canada only)Fax: (734) 971-3640