News & Announcements
Growth Factor Receptors in Cancer
Growth Factor Receptors in Cancer
Article from 2021-04-01
Growth factor signaling regulates numerous cellular processes, including differentiation, survival, migration, and proliferation. Overactivation of these signaling pathways resulting from amplification, overexpression, or mutations in growth factor receptors occurs in a variety of cancers. These alterations to growth factor signaling pathways contribute to cancer initiation, progression, angiogenesis, and metastasis. Several FDA-approved cancer therapeutics modulate growth factor signaling by inhibiting the activity of growth factor receptors. As cancer cells develop resistance mechanisms to growth factor receptor inhibition, further drug development will be needed to overcome these challenges.
Epidermal Growth Factor Receptors
The EGFR family consists of four receptor tyrosine kinases, namely EGFR/HER1/ErbB1, HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4. EGFR family signaling has roles in cell survival, differentiation, motility, adhesion, and proliferation, as well as angiogenesis.1,2 In canonical signaling, ligand binding to the extracellular domain of EGFRs, except for that of HER2, induces either homo- or heterodimerization of EGFR family receptors. Activated EGFRs then phosphorylate various target proteins to activate downstream pathways including the PI3K/Akt, RAS/RAF/MEK/ERK, and phospholipase c (PLC)/PKC pathways, which are common among many receptor tyrosine kinases (Figure 1).
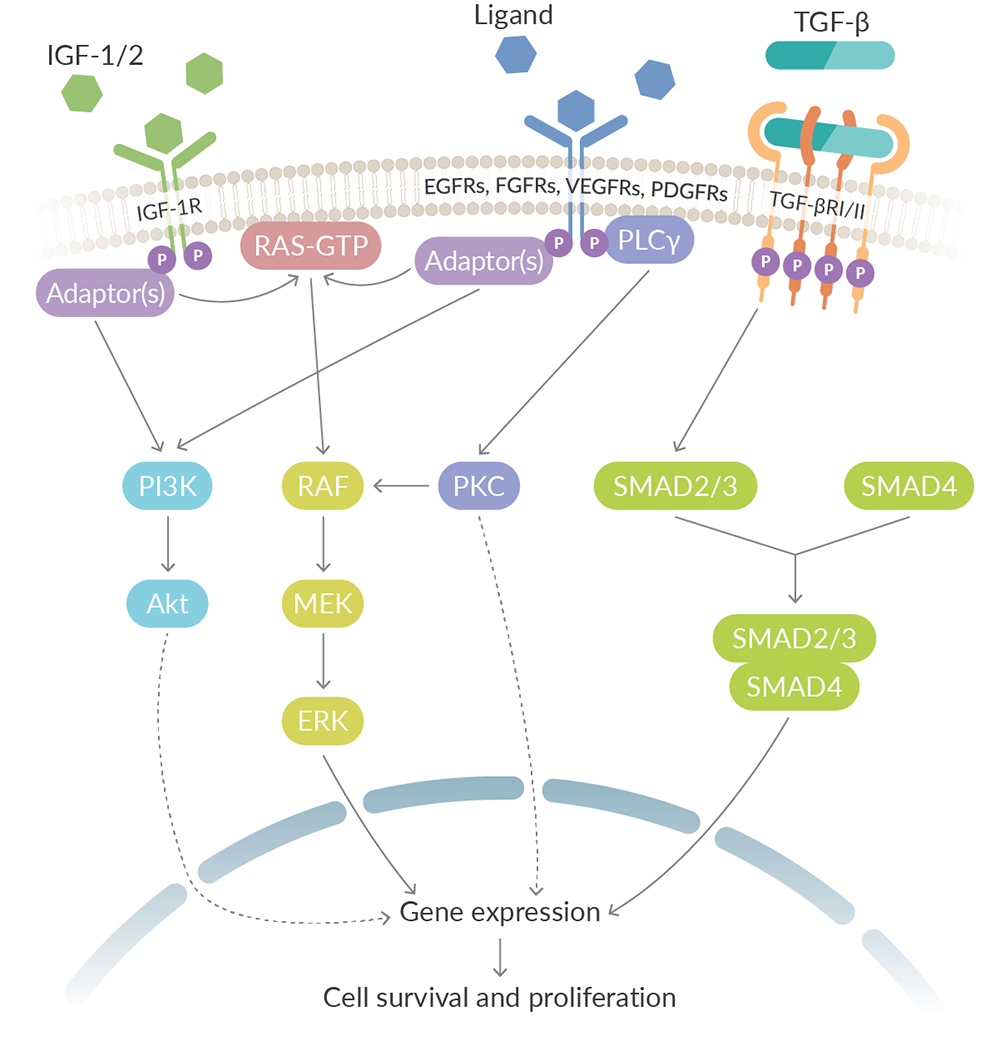
Figure 1. Activation of canonical growth factor signaling pathways by ligand binding promotes cell survival and proliferation.
Dysregulation of EGFR signaling is frequently associated with cancer and can be caused by mutations within the genes encoding EGFR family members, gene amplification, and/or protein overexpression.1EGFR is amplified and/or overexpressed in patients with breast, bladder, or lung cancer and is mutated and/or amplified in non-small cell lung cancer (NSCLC) and gliomas.3,4 Activating mutations in the catalytic domain of EGFR include in-frame deletions in exon 19 and the L858R mutation in exon 21.4 Another common mutation in EGFR is the T790M mutation, which confers resistance to certain kinase inhibitors.
Overexpression of HER2 occurs in up to 30% of breast cancers and is also amplified and/or overexpressed in NSCLC and gastric, bladder, or ovarian cancers.2,4 Mutations in HER2 have been found in breast, colorectal, and bladder cancers, with the most common mutations occurring in the extracellular and kinase domains.4 The S310F/Y mutation, which occurs in the extracellular domain, is most commonly found in bladder cancer, while kinase domain mutations, including L755S, V777L, V842I, and exon 20 insertions/deletions, have been found in breast, colorectal, and lung cancers.
Mutations in HER3 and HER4 are less frequent. Mutations in HER3 have been found in breast, gastric, ovarian, colon, and head and neck cancers, as well as glioblastoma and squamous cell carcinomas. The V855A mutation in the HER3 kinase domain, which is homologous to the EGFR L858R mutation, has been found in patients with NSCLC. Mutations in HER4 have been found in melanoma and NSCLC, among others.
Numerous EGFR inhibitors have been approved by the FDA for the treatment of cancers with aberrant EGFR family signaling and are the standard first-line treatment for advanced NSCLC.5 The EGFR inhibitors gefitinib and erlotinib are both approved for the treatment of NSCLC in patients bearing EGFR exon 19 deletions or L858R mutations. A third-generation kinase inhibitor, osimertinib (AZD 9291), inhibits EGFR bearing the T970M mutation, overcoming acquired resistance created by T970M mutations in NSCLC. Other EGFR family inhibitors approved by the FDA for the treatment of NSCLC include afatinib and dacomitinib (PF-299804), which both inhibit EGFR, HER2, and HER4. Lapatinib, a dual inhibitor of EGFR and HER2, is approved for combination therapy in patients with HER2-overexpressing metastatic breast cancers, and neratinib is approved as a single agent or in combination therapy for HER2-positive breast cancer. Cayman offers inhibitors of wild-type and mutant EGFRs to study EGFR signaling and inhibitor resistance in cancer cells.
See all EGFR family inhibitors
Vascular Endothelial Growth Factor Receptors
VEGFRs are transmembrane receptor tyrosine kinases with roles in cell proliferation and migration, angiogenesis, lymphangiogenesis, and immune cell recruitment.3,6 Activation of VEGFR1 (FLT1), which is primarily expressed in vascular endothelial cells, stimulates the migration of monocytes and macrophages.3,7 VEGFR2 (KDR/FLK1), which is also primarily expressed in vascular endothelial cells, stimulates cell migration, induces MAPK-dependent proliferation, and regulates angiogenesis.3,6,7 The third member of this family, VEGFR3 (FLT4), is expressed in lymphatic endothelial cells and regulates lymphangiogenesis.6,7
Increased expression of VEGFRs has been observed in a variety of cancer types, including gastric, colon, bladder, brain, breast, lung, ovarian, and prostate cancers, as well as head and neck carcinomas.3 Upregulation of VEGFR2 occurs in the vascular endothelium of most common solid tumor types and increased levels of VEGFR2 in tumor tissue from patients with gastric cancer are associated with poor prognosis.6,8 Somatic mutations in VEGFR2 and VEGFR3 have also been found in patients with juvenile hemangioma.3
Several FDA-approved kinase inhibitors target VEGFR signaling. However, most of these also have additional targets. Sunitinib, which is approved for the treatment of gastrointestinal stromal tumors (GISTs), renal cell carcinoma, and pancreatic neuroendocrine tumors, inhibits VEGFR1-3, as well as PDGFRα, PDGFRβ, KIT, and RET.9Sorafenib inhibits VEGFR1-3, PDGFRβ, KIT, RET, B-RAF, C-RAF, and FLT3 and is used to treat hepatocellular, renal cell, and differentiated thyroid carcinomas. Pazopanib is approved for the treatment of renal cell carcinoma and soft tissue sarcoma and inhibits VEGFR1-3, PDGFRα, PDGFRβ, FGFR1, FGFR3, and KIT. Axitinib targets VEGFR1-3 and PDGFRβ and is used as a single agent or in combination therapy for renal cell carcinoma. Numerous additional VEGFR inhibitors are available from Cayman to study VEGFR-dependent signaling and angiogenesis in the tumor microenvironment.
See all VEGFR family inhibitors
Fibroblast Growth Factor Receptors
The FGFR family of receptor tyrosine kinases consists of FGFR1-4. Signaling through FGFRs activates downstream signaling through the MAPK, PI3K/Akt, and PLC/PKC pathways to regulate cellular differentiation, proliferation, survival, and migration, as well as angiogenesis, vascular repair, and wound healing (Figure 1).3,10
Translocation, amplification, and/or activating mutations in FGFR genes have been found in a variety of cancers, including gastric, lung, breast, and ovarian cancers.3,10 Fusion genes between FGFR1-3 and a variety of partners have been found in glioblastoma, cholangiocarcinoma, NSCLC, and urothelial bladder carcinoma.10 Amplification of FGFR1 has been found in osteosarcoma, squamous NSCLC, small-cell lung cancer, and breast cancer. Amplification of FGFR2 in triple-negative breast cancer and gastric cancers and overexpression of FGFR4 in breast cancer are associated with poor prognosis.2,10 Activating mutations in FGFR2, FGFR3, and FGFR4 have been found in endometrial cancer, urothelial bladder carcinoma, and rhabdomyosarcoma, respectively.10
Erdafitinib (JNJ-42756493) and pemigatinib are FDA-approved FGFR inhibitors.9,11 Erdafitinib inhibits FGFR1-4 and is approved for the treatment of urothelial carcinoma with susceptible FGFR2 or FGFR3 genetic alterations, whereas pemigatinib inhibits FGFR1-3 and is used to treat cholangiocarcinoma with FGFR2 fusions or rearrangements. Cayman offers additional FGFR inhibitors to study FGFR signaling in cancer cells.
See all FGFR family inhibitors
Insulin-Like Growth Factor Receptors
There are two insulin-like growth factor (IGF) receptors: IGF-1R and IFG-2R. IGF-1R is a receptor tyrosine kinase, whereas IGF-2R lacks a kinase domain.2 Signaling through IGF-1R, which is induced by binding of IGF-1 or IGF-2, regulates cell growth, proliferation, and motility through activation of the MAPK and PI3K signaling pathways (Figure 1). Because IGF-2R lacks a kinase domain, it cannot transduce signals. Instead, IGF-2R removes the ligand IGF-2 through receptor-mediated endocytosis and subsequent degradation.12
Amplification of IGF1R has been found in melanoma and breast cancer, with increased levels of IGF-1R in almost 80% of breast cancers compared with non-cancerous breast tissue.2,3 Mutations in IGF2R have been found in lung squamous cell carcinomas.3
There are currently no FDA-approved small molecule inhibitors of IGFRs. Two FDA-approved tyrosine kinase inhibitors that do target IGF-1R in some fashion are brigatinib (AP26113) and ceritinib, which are both approved for the treatment of anaplastic lymphoma kinase-positive (ALK+) NSCLC. However, IGF-1R is not the primary target for either of these inhibitors. Instead, both primarily target ALK.9 Cayman has numerous IGF-1R inhibitors available to aid in preclinical and drug discovery studies.
Platelet-Derived Growth Factor Receptors
PDGFR signaling regulates cell growth, proliferation, differentiation, survival, and migration, including the growth and differentiation of mesenchymal stem cells, and has roles in angiogenesis.3,13 Ligand binding causes either homo- or heterodimerization of PDGFRα and PDGFRβ and induces activation of downstream signaling pathways, including the MAPK, PI3K/Akt, and PLCγ pathways (Figure 1).13,14
Amplification of PDGFRA, which encodes PDGFRα, has been found in patients with glioblastoma, and activating mutations in PDGFRα have been found in GISTs.14 Gain-of-function mutations in PDGFRB, which encodes PDGFRβ, have been found in myofibromatosis, and genetic rearrangements leading to expression of fusion proteins between PDGFRβ and the transcription factor Tel have been found in patients with chronic myelomonocytic leukemia.3,14 In prostate cancer, expression of PDGFRβ in the stroma of tumors and adjacent non-malignant tissue is used as a prognostic marker.13
Avapritinib (BLU-285) is an inhibitor of PDGFRα bearing the activation loop mutation D842V, as well as several mutant forms of KIT.15 It is FDA approved for the treatment of GISTs harboring PDGFRα exon 18 mutations, including the PDGFRα D842V mutation. Ripretinib inhibits both wild-type and D842V mutant PDGFRα, as well as wild-type, oncogenic, and drug-resistant mutants of KIT.16 It also inhibits PDGFRβ, VEGFR2, Tie2, and DDR2, and is approved by the FDA for the treatment of GISTs. Many kinase inhibitors that target PDGFRs also hit additional targets because the ATP binding pocket is conserved across tyrosine kinases.13 Cayman offers many additional PDGFR inhibitors to study the role of this signaling pathway in cancers.
See all PDGFR family inhibitors
TGF-β Receptors
TGF-β signaling through TGF-β receptors (TGF-βRs) regulates cell growth, proliferation, differentiation, survival, and migration, as well as angiogenesis.2,3 Unlike the previously discussed growth factor families, the TGF-βRs are not receptor tyrosine kinases. Instead, TGF-βRI (TβRI/ALK5) and TGF-βRII (TβRII) are transmembrane receptors with serine/threonine kinase activity, whereas TGF-βRIII (TβRIII) lacks the kinase domain.3 In canonical signaling, binding of TGF-β to TGF-βRII leads to the recruitment of TGF-βRI and downstream signaling through SMAD2, SMAD3, and SMAD4 (Figure 1).2
In cancer, TGF-β signaling can act as either a tumor suppressor or a promoter of tumor progression, depending on the context.2,17 In healthy tissue or in the early stages of tumor formation, TGF-β can inhibit cell cycle progression and cell proliferation while inducing apoptosis, thereby limiting tumor progression. At later stages of tumorigenesis, however, TGF-β can promote proliferation and invasion of cancer cells, as well as metastasis, due to genetic and epigenetic changes. 2,3,17
Mutations in TGFBR1, the gene encoding TGF-βRI, have been found in patients with breast, ovarian, and pancreatic cancers, as well as patients with T cell lymphoma.17 Mutations in TGFBR2, which encodes TGF-βRII, have been found in patients with colon and pancreatic cancers, and down-regulation of the gene has been observed in breast and lung cancers.3
While several TGF-βR inhibitors have been explored at the preclinical and clinical levels for cancer treatment, none are currently FDA approved. One such compound, galunisertib (LY2157299), inhibits TGF-βRI and showed promise in in vitro and in vivo models of hepatocellular carcinoma, but its development by Eli Lilly was discontinued in January 2020.2,17 Cayman offers TGF-βR inhibitors to study TGF-β signaling in physiological and pathological contexts.
Therapeutic Challenges
While several growth factor receptor inhibitors have been approved by the FDA for cancer treatment, there are still challenges to this therapeutic approach in the form of intrinsic or acquired resistance.3,9 Mutations within receptors can confer resistance by preventing inhibitor binding. Cells can also overcome receptor inhibition by overexpressing the targeted receptor. Outside of alterations to the receptors themselves, cells can compensate for receptor inhibition by activating downstream or parallel signaling, with the latter mechanism taking advantage of the natural crosstalk between pathways. For example, overexpression of FGFRs can contribute to acquired resistance to inhibitors of EGFR, VEGFR, and PDGFR family members, due to extensive cross-talk between these pathways and FGFR signaling (Figure 1).3 Resistance to these therapeutics can also be achieved through the expression of efflux pumps that transport the inhibitors out of the cells.
Continued development of growth factor receptor inhibitors, either as single agents or in combination therapies, will help to overcome these challenges. Cayman offers a variety of tools and services to study growth factor receptor signaling and inhibition in both physiological and pathological contexts.
Explore our large collection of kinase inhibitors, many of which have been organized into the ~160-compound Kinase Screening Library (96-Well) and the ~850-compound Comprehensive Kinase Screening Library to simplify your screening efforts.
Cayman offers several cell-based reporter assay kits from INDIGO Biosciences to measure growth factor receptor signaling and aid in the screening and development of new therapeutics targeting EGFR, VEGFR2, or TGF-βRI/II. These assay kits are compatible with both 96- and 384-well formats, allowing for screening of a large number of samples.
Human Epidermal Growth Factor Receptor 1 Reporter Assay System
Human Vascular Endothelial Growth Factor Receptor 2 Reporter Assay System
Human Transforming Growth Factor Beta Receptors I/II Reporter Assay System
Cayman also provides custom synthesis services and structure-based drug design services to assist you with SAR studies.
You May Also Be Interested In
 Methods for Detecting Kinase Activity |  Small Molecule Inhibitors Selection Guide |
 Cancer |  Download Our Kinase Pathway Chart |
References:
1. Sigismund, S., Avanzato, D., and Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 12(1), 3-20 (2018).
2. Du, J., Yu, Y., Zhan, J., et al. Targeted therapies against growth factor signaling in breast cancer. Translational research in breast cancer. Song, E. and Hu, H., editors, Springer (2017).
3. Tiash, S. and Chowdhury, E.H. Growth factor receptors: Promising drug targets in cancer. J. Cancer Metastasis Treat. 1(3), 190-200 (2015).
4. Mishra, R., Hanker, A.B., and Garrett, J.T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget 8(69), 114371-114392 (2017).
5. Solassol, I., Pinguet, F., and Quantin, X. FDA- and EMA-approved tyrosine kinase inhibitors in advanced EGFR-mutated non-small cell lung cancer: Safety, tolerability, plasma concentration monitoring, and management. Biomolecules 9(11), 668 (2019).
6. Rapisarda, A. and Melillo, G. Role of VEGF/VEGFR axis in cancer biology and therapy. Adv. Cancer Res. 114, 237-267 (2012).
7. Ivy, S.P., Wick, J.Y., and Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 6(10), 569-579 (2009).
8. Lian, L., Li, X.-L., Xu, M.-D., et al. VEGFR2 promotes tumorigenesis and metastasis in a pro-angiogenic-independent way in gastric cancer. BMC Cancer 19(1), 183 (2019).
9. Pottier, C., Fresnais, M., Gilon, M., et al. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers (Basel) 12(3), 731 (2020).
10. Touat, M., Ileana, E., Postel-Vinay, S., et al. Targeting FGFR signaling in cancer. Clin. Cancer Res. 21(12), 2684-2694 (2015).
11. Liu, P.C.C., Koblish, H., Wu, L., et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One 15(4), e0231877 (2020).
12. Cao, J. and Yee, D. Disrupting insulin and IGF receptor function in cancer. Int. J. Mol. Sci. 22(2), 555 (2021).
13. Papadopoulos, N. and Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Aspects Med. 62, 75-88 (2018).
14. Kazlauskas, A. PDGFs and their receptors. Gene 614, 1-7 (2017).
15. Evans, E.K., Gardino, A.K., Kim, J.L., et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 9(414), eaao1690 (2017).
16. Smith, B.D., Kaufman, M.D., Lu, W.-P., et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell 35(5), 738-751 (2019).
17. Gómez-Gil, V. Therapeutic implications of TGFβ in cancer treatment: A systematic review. Cancers (Basel) 13(3), 379. (2021).
Contact Info
Cayman Chemical1180 East Ellsworth RoadAnn Arbor, Michigan 48108 USAToll Free: (800) 364-9897(USA and Canada only)Fax: (734) 971-3640