News & Announcements
Casting NETs in COVID-19
Article from 2020-06-25
Neutrophils, well known for their role in the engulfment and destruction of invasive pathogens, have now been blamed for the exacerbated host response in patients with severe COVID-19. Their aberrant activation is linked to increased pulmonary inflammation and elevated levels of serum pro-inflammatory cytokines that lead to extensive lung damage and thrombosis. Moreover, excessive circulating neutrophils (neutrophilia), outnumbering existing T cells, B cells, and natural killer cells, predicts poor outcomes in patients with COVID-19.
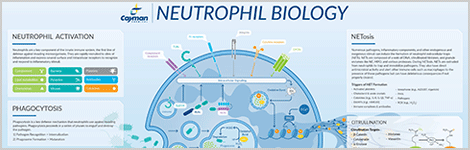
Neutrophil Biology
When an infection occurs, neutrophils are rapidly mobilized and recruited through a three-step process. First, selectin-mediated rolling adhesion to the vascular endothelial cells slows the progression of the neutrophil in the circulation. Next, integrin-mediated firm adhesion promotes the arrest of forward motion. Finally, transendothelial migration allows the neutrophil to leave the circulation and crawl into the underlying infected tissue. Chemoattractants, such as leukotriene B4, act as homing signals to guide the extravasated neutrophils directly to the inflammatory site. Once the neutrophil finds its way to the site of infection, its encounter with the pathogen is mediated by one or more pattern recognition receptors (PRRs) that bind to specific pathogen-associated molecular patterns (PAMPs) common to invasive microorganisms. The interaction of PRRs with PAMPs promotes the initiation of signal transduction cascades that trigger the activation of neutrophil antimicrobial defenses. The pathogen is killed by oxidative burst, the release of neutrophil extracellular traps (NETs), and phagocytosis. Once this arsenal has been dispensed, neutrophils typically undergo apoptosis to prevent further damage to surrounding tissue. They are efficiently phagocytosed by macrophages that follow neutrophils into the inflammatory site.
NETs
Almost every infectious challenge induces NETosis, the process whereby neutrophils extrude a web of DNA, histones, and antimicrobial macromolecules (e.g., myeloperoxidase (MPO), neutrophil elastase (NE), defensins, etc.) (Figure 1). The exact mechanism of NET formation remains unclear, but intermediate steps require reactive oxygen (ROS) species generated from the actions of NADPH oxidase. NE from the primary granules within the neutrophil migrates to the nucleus where it degrades the linker histones (histone H1). MPO also migrates to the nucleus where it enhances the process of chromatin decondensation. The nuclear enzyme peptidylarginine deiminase 4 (PAD4) is also engaged. PAD4 deiminates the chromatin core histones (H2A, H2B, H3, and H4), reducing their binding affinity for DNA, and promoting the unwinding of DNA from the core histones. It does so by catalyzing the post-translational modification of arginine to citrulline within histones, which disrupts ionic interactions and promotes chromatin decondensation. Gasdermin D generates pores in the membrane of the neutrophil, causing the membrane to rupture and expel its contents. Through these actions, a meshwork of DNA and histones is woven together with antimicrobial enzymes and cast from the cytoplasm into the extracellular space. The NET traps the infectious pathogen and promotes phagocytosis by inflammatory macrophages, facilitating the indirect killing of these pathogens. The spent NET is digested by serum DNase 1, and the remaining particles are removed by scavenger receptor-mediated phagocytosis by macrophages.
Collateral Damage
Although NETs are part of the host's innate immune response, excessive NET formation can trigger numerous inflammatory reactions that promote cancer cell metastasis, destroy surrounding tissues, facilitate thrombosis, and result in permanent organ damage to the pulmonary, cardiovascular, and renal systems. Aberrant NET formation has been linked to acute respiratory distress syndrome, as neutrophils in cases associated with pneumonia seem primed to produce NETs.1 The mucous secretions that impair gas exchange in cystic fibrosis have been shown to contain extracellular DNA derived from NETs released in response to lung infections.2 These secretions are thicker and more viscous from the increased presence of NE, which only further promotes problems with respiration and an uptick in secondary infections that leads to the recruitment of more neutrophils.
Intravascular NETs play a crucial role in promoting thrombosis in arteries and veins by activating the contact pathway of coagulation (Figure 2). Platelets activated through a process dependent upon thromboxane A2 adhere to neutrophils and induce NET formation.3 Histones released from NETs promote thrombin generation and platelet aggregation by acting as ligands for Toll-like receptors on platelets.4 NE released from NETs may also play a role in digesting coagulation inhibitors. A feedback cycle is initiated whereby pro-coagulant activity from thrombin leads to additional platelet activation that then further enhances NET formation. When NETs circulate at high levels in blood, their activity can trigger excess clotting and occlude small vessels. Neutrophilia is an associated predisposition to thrombosis and predicts poor outcomes in patients with COVID-19. Indeed, extensive neutrophil infiltration and inflammation in the capillaries of the lungs as well as neutrophils in the alveoli of the lungs have been identified in autopsy specimens from patients who succumbed to COVID-19.5 Sera from patients with COVID-19 have elevated levels of two specific markers of NETs: myeloperoxidase and citrullinated histone H3.6 Thus, NET activity may likely contribute to the lung injury and thrombosis associated with severe COVID-19.
NET Therapeutics
Targeting intravascular NETs may reduce thrombosis and lung injury in patients with severe COVID-19. While preventing platelet activation could indirectly lead to reduced NET formation, inhibiting the enzymes essential to NET formation such as PAD4 and NE would directly block their assembly. Cayman has developed several tools to identify NETs and inhibit their formation as well as compounds to control platelet activation.
You May Also Be Interested In
| | |

References
1. Adrover, J.M., Aroca-Crevillén, A., Crainiciuc, G., et al. Programmed 'disarming' of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 21(2), 135-144 (2020).
2. Martínez-Alemán, S.R., Campos-García, L., Palma-Nicolas, J.P., et al. Understanding the entanglement: Neutrophil extracellular traps (NETs) in cystic fibrosis. Front. Cell. Infect. Microbiol. 7, 104 (2017).
3. Caudrillier, A., Kessenbrock, K., Gilliss, B.M., et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122(7), 2661-2671 (2012).
4. Semeraro, F., Ammollo, C.T., Morrissey, J.H., et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 118(7), 1952-1961 (2011).
5. Barnes, B.J., Adrover, J.M., Baxter-Stoltzfus, A., et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 217(6), e20200652 (2020).
6. Zuo, Y., Yalavarthi, S., Shi, H., et al. Neutrophil extracellular traps in COVID-19. JCI Insight 5(11), 138999 (2020).
Contact Info
Cayman Chemical1180 East Ellsworth RoadAnn Arbor, Michigan 48108 USAToll Free: (800) 364-9897(USA and Canada only)Fax: (734) 971-3640